10.3.11 Určení aktivitního koeficientu a aktivit složek z dat o rovnováze
V následující tabulce jsou uvedeny experimentálně určené hodnoty p−x1−y1 platné pro systém ethanol(1) - voda(2) při 70 °C.
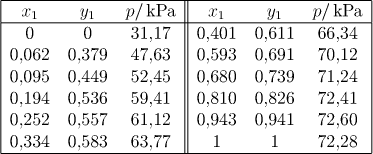
Rovnovážná data ![]() v systému ethanol(1)–voda(2)
při teplotě 70 °C.
v systému ethanol(1)–voda(2)
při teplotě 70 °C.
a) aktivitní koeficienty obou složek,
b) aktivity obou složek,
c) μi−μ•i obou složek.
Do grafu ai = f(x1) a μi−μ•i = f(x1) zakreslete též závislost, která odpovídá ideálnímu roztoku (μ•i jsou molární Gibbsovy energie čistých látek v kapalném stavu) za teploty a tlaku soustavy. Je možno k vystižení závislosti aktivitních koeficientů na složení použít vztah
![]()
platný pro regulární roztok?
↓
↓
↓
↓
↓
↓
↓
↓
↓
↓
↓
↓
↓
Řešení
V případě rovnováhy kapalina–pára počítáme aktivitní koeficienty ze vztahu
![]()
Např. pro x1 = 0,062 získáme
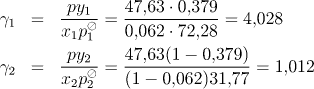
Aktivity obou složek určíme ze vztahu
![]()
Pro x1 = 0,062 obdržíme
![]()
Pro chemický potenciál složky i platí relace
![]()
z níž pro x1 = 0,062 dostaneme
![]()
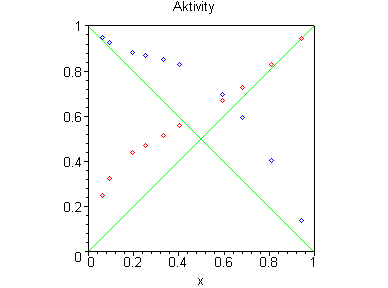
Výsledky pro zbývající složení jsou uvedeny v následující tabulce a obrázku.
| x1 | γ1 | γ2 | a1 | a2 | (μ1−μ•1) | (μ2−μ•2) |
| 0 | (5,36) | 1 | 0 | 1 | −∞ | 0 |
| 0,062 | 4,028 | 1,012 | 0,250 | 0,949 | -3957,9 | -149,5 |
| 0,095 | 3,430 | 1,024 | 0,326 | 0,927 | -3199,4 | -215,7 |
| 0,194 | 2,271 | 1,097 | 0,441 | 0,884 | -2338,6 | -350,5 |
| 0,252 | 1,869 | 1,161 | 0,471 | 0,869 | -2148,0 | -401,7 |
| 0,334 | 1,540 | 1,281 | 0,514 | 0,853 | -1896,7 | -453,2 |
| 0,401 | 1,398 | 1,382 | 0,561 | 0,828 | -1650,2 | -538,7 |
| 0,593 | 1,130 | 1,708 | 0,670 | 0,695 | -1141,1 | -1037,5 |
| 0,680 | 1,071 | 1,864 | 0,728 | 0,597 | -904,2 | -1473,9 |
| 0,810 | 1,022 | 2,127 | 0,827 | 0,404 | -540,2 | -2584,2 |
| 0,943 | 1,002 | 2,411 | 0,945 | 0,137 | -160,9 | -5662,3 |
| 1 | 1 | (2,51) | 1 | 0 | 0 | −∞ |
Vypočítané hodnoty aktivitních koeficientů, aktivit a
μi−μ•i (v J/mol) u systému ethanol(1) -
voda(2) při 70 °C.
Jednotlivé závislosti můžeme znázornit také v grafech.
![[Maple Plot]](images/fazVIII7.gif)
![[Maple Plot]](images/fazVIII8.gif)
![[Maple Plot]](images/fazVIII9.gif)